

INTRODUCTION
Myositis ossifican (MO) is commonly characterised as a benign, self-limiting ossifying lesion in extraskeletal soft tissues (Cherry et al. 2023). It is considered a rare type of heterotrophic ossificans that involves bone formation in soft tissue sites, including skeletal muscle, subcutaneous fat, tendon and nerve (W. T. Li, Horng, and Chien 2016). There are 3 main sub categories of myositis ossificans: fibrodysplasia ossificans progressiva (severe systemic form of heterotrophic ossificans), pseudomalignant/atraumatic MO (without a history of trauma) and circumscribed MO (with a background of trauma) (Rehman et al., n.d.). Clinically, MO can be initially misdiagnosed as malignancy (commonly osteosarcoma) due to its inherent similarities in the patient’s physical and radiographic findings.
We present a case of MO of the clavicle in a 13 year old boy who did not have any significant history of preceding trauma. Although MO can present in any ages, it is most commonly found in those aged 20 to 30 and is known to be rare in the paediatric population (Saad et al. 2021). MO also commonly presents in the extremities instead of the head and neck region, making our case exceedingly rare (Cherry et al. 2023). To the best of our efforts in reviewing existing literature, there have been no previously published case reports of atraumatic MO of the clavicle in a paediatric patient.
CASE REPORT
The 13-year-old school boy came with complains of a fast growing, painful mass over the medial end of the left shoulder over the last 2 months. He had difficulty achieving a full range-of-motion of the shoulder (mainly abduction) due to pain. He also complained of night pain which occasionally wakes him up at night. He had no history of fever or sudden loss of weight and appetite. He had also no known history of trauma, although we cannot exclude the possibility of any inciting event (ie secondary to a heavy school bag, physical activities in school). He had no significant past medical history.
CLINICAL FINDINGS
Upon inspection, there was an 8x7cm diffuse swelling over the medial half of the left clavicle. Skin over the clavicle was warm to touch and tender, with mild discoloration. Mild erythema was also noted. There was no palpable cervical or axillary lymph nodes and abdominal examination was unremarkable.
DIAGNOSTIC ASSESSMENT
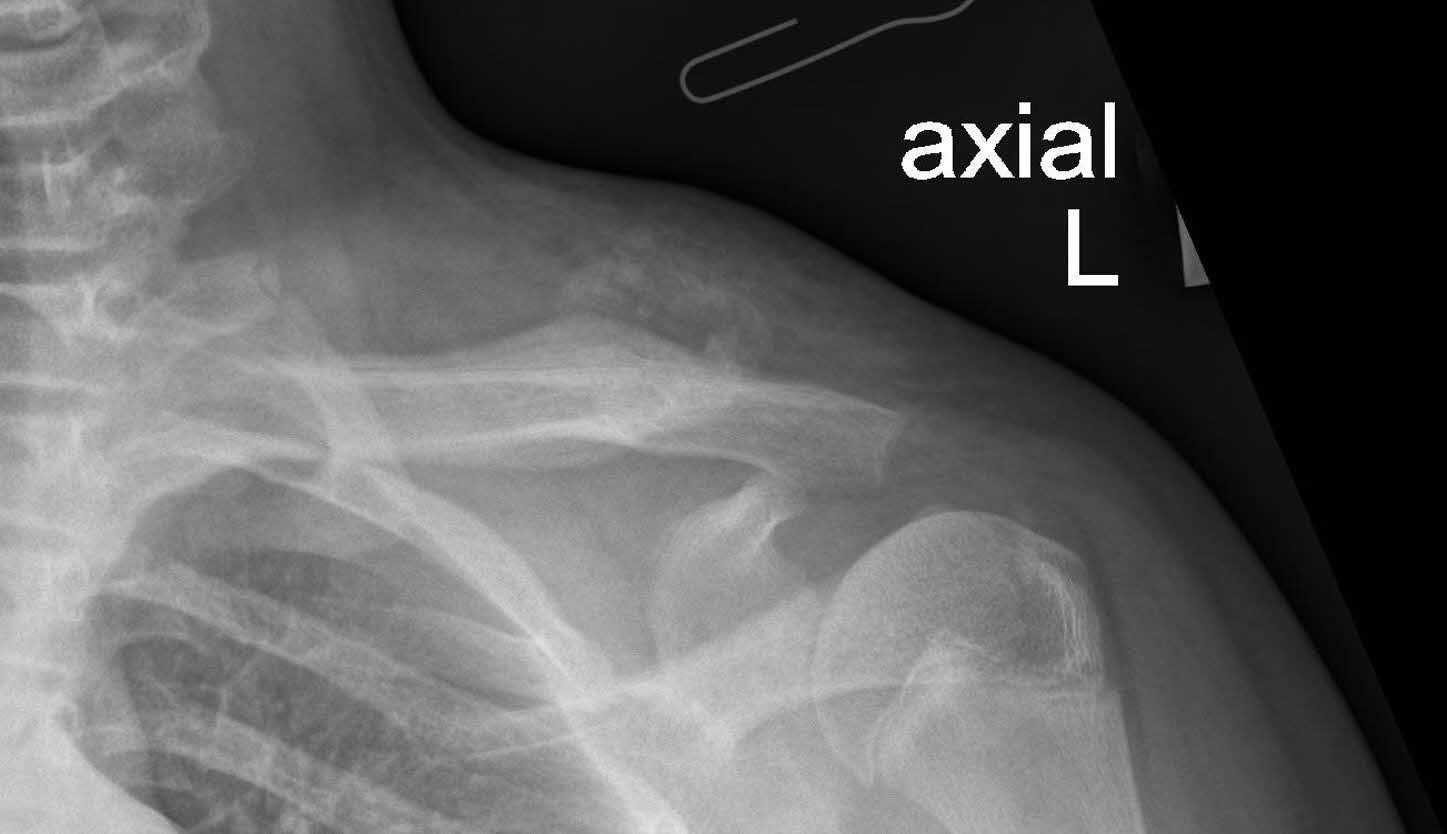
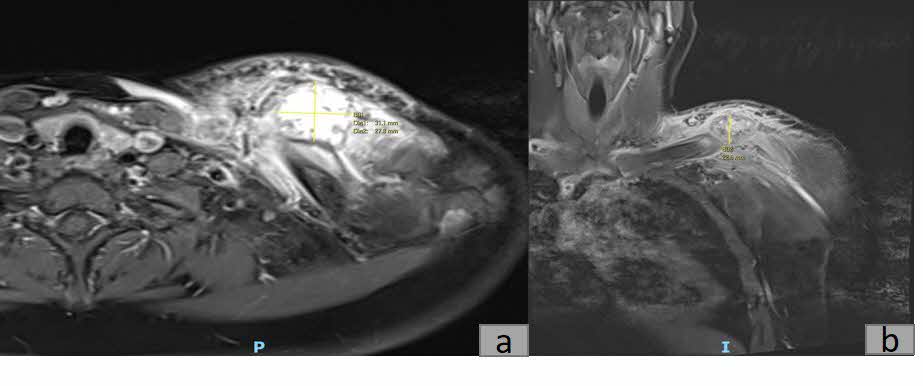
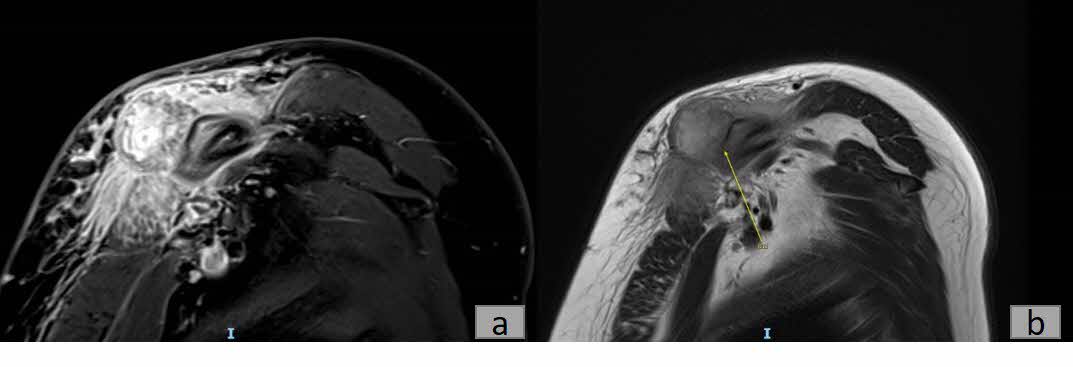
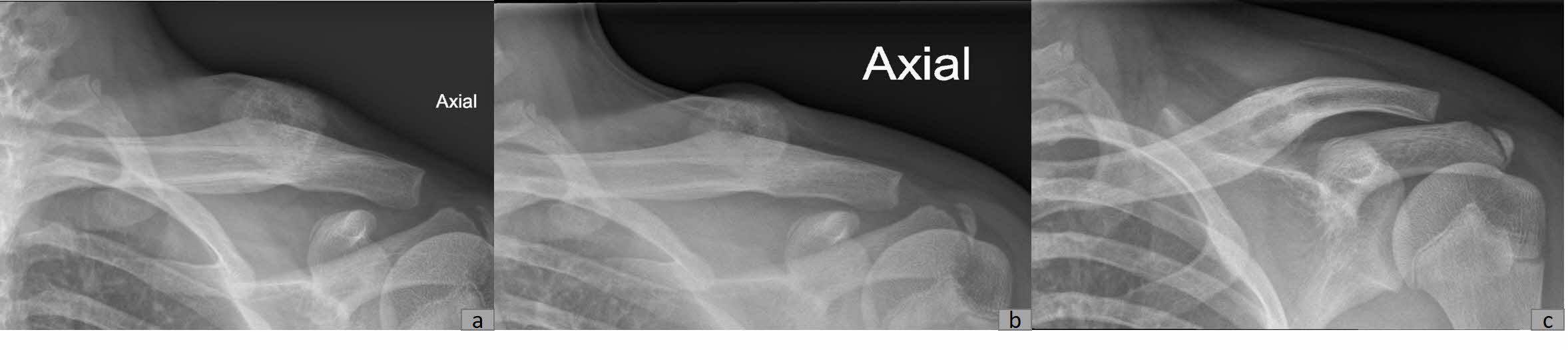
Plain radiography revealed a calcified mass with associated periosteal reaction along the left clavicular shaft (Figure 1). An MRI scan was also done and reported an avidly-enhancing T1w-isointense and T2w-hyperintense lesion in the middle-distal third of the left clavicle, measuring approximately 3.1cm x 2.8cm x 2.3cm (Figure 2). In certain areas, the lesion was inseparable from the adjacent periosteum (Figure 3), which itself was significantly thickened and enhancing. There was also increased T2W marrow edema with enhancement and some adjacent soft tissue swelling. No associated fracture was noted. The MRI report was suggestive of a malignant lesion such as soft tissue sarcoma.

_and_coronal_(b)_t1_weighted_magnetic_resonance_images_of_a_3.1cm_x_2.8cm_x_2.3cm.jpg)
_and_t2_fblade_mri_(b)_showing_that_the_lesion_i.jpg)
Incisional biopsy and molecular gene testing were performed. Microscopic analysis of the biopsy revealed multiple fragments with variable cellularity, with some showing features of maturation. Some fragments showed plump stromal cells with bland nuclear features and a background of fibrocollagenous to hyalinised stroma with some osseous debris. There were trabeculae of woven bone bordered by plump osteoblasts with occasional osteoclasts and a fibrinous exudate. No significant cytological atypia was noted. In addition, molecular testing identified a COL1A1::USP6 gene fusion (via Archer FusionPlex Anchored multiplex PCR). Interphase fluorescence in situ hybridization (FISH) for MDM2 (12q15) was also performed and this test was negative for MDM2 gene amplification.
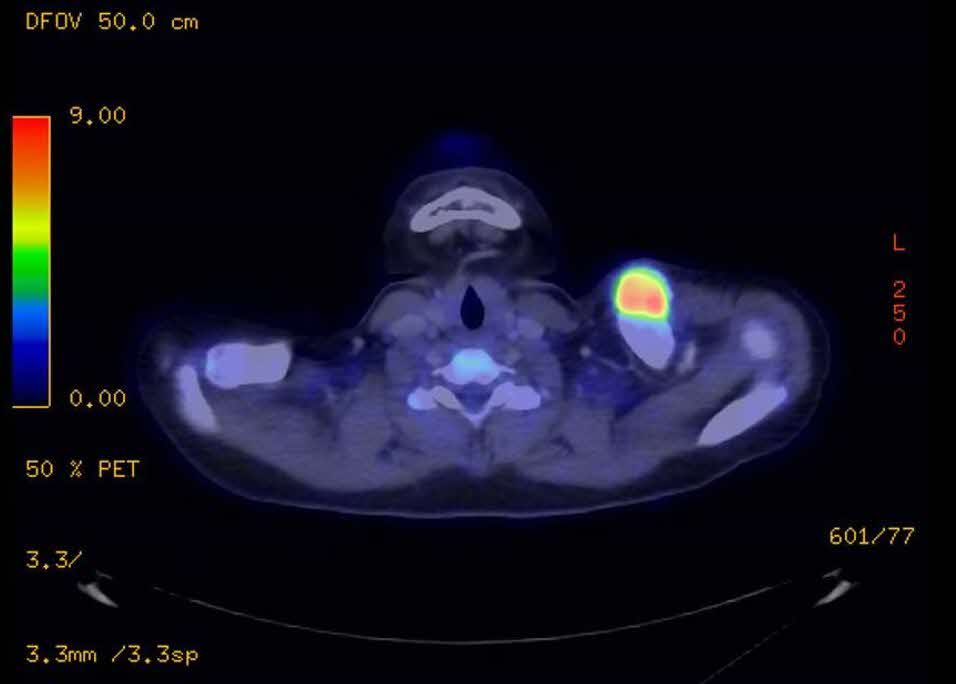
While awaiting the histopathological report, a PET/CT scan was also performed to look for potential metastasis. Report from the PET/CT scan (Figure 4) showed a markedly FDG-avid bone forming lesion (SUV MAX value: 9.7) in the left clavicle, with no other FDG-avid lesion detected in the body.

THERAPEUTIC INTERVENTION AND FOLLOW UP
Subsequently, the patient was referred to musculoskeletal oncology given the high suspicion of malignancy. Conservative treatment was adopted for this patient, including a 3 months prescription of Indomethacin and continual radiographic monitoring. Spontaneous resolution of the mass was observed over the next six months, as evidenced by plain radiography (Figure 5). At the fifth month of follow-up, the patient was asymptomatic and exhibited normal function with a full range of motion in the left shoulder. Consequently, the patient was given an open dated review.
_of_the_left_shoulder_taken_2_months_(a)__3_months_(b)_and_6_month.jpg)
DISCUSSION
The exact pathogenesis of MO is not well understood in literature. In cases of circumscribed MO preceded by a history of trauma, it is hypothesized that trauma caused an inappropriate differentiation of fibroblasts into osteogenic cells (Walczak, Johnson, and Howe 2015). Some studies have suggested that Endothelial-mesenchymal transition could contribute to the development of MO. Inflammatory cytokines (bone morphogenetic protein-2 & -4) cause endothelial-derived mesenchymal stem cells to differentiate into chondrocytes or osteoblasts, resulting in bone formation in soft tissue (Walczak, Johnson, and Howe 2015).
However, pathogenesis of atraumatic MO remains unclear. Atraumatic MO is significantly rarer than circumscribed MO (Rehman et al., n.d.). Clinically, atraumatic MO can mimic bone and soft tissue malignancy, and plain radiography is insufficient to diagnose MO.
The process of MO can be grouped into 3 stages: early (first 4 weeks), intermediate (4- 8 weeks), and mature (months later). Calcification typically occurs during the intermediate stage, while mature bone formation develops in the mature stage. Bone tumours, like osteosarcoma, can also exhibit calcification and bone formation. Diagnosis of MO by plain radiography is generally possible during the mature stage, when the characteristic features, such as bone formation with central lucency, become apparent.
However, in the early stage of MO, radiography and MRI cannot fully confirm the diagnosis of MO and exclude the possibility of malignancy. Delaying diagnosis by waiting for the tumour to mature is not advisable, as such delays in treatment – particularly if the tumour turns out to be osteosarcoma – can have severe consequences and poor prognosis. In this case, the patient presented to us 2 months after first noticing the growing mass in his clavicle. Given the duration, the tumour was likely not yet mature, rendering MRI and radiography inconclusive. Consequently, biopsy and molecular testing were therefore indicated in this case with diagnostic uncertainty.
The main differential diagnoses being considered were Parosteal osteosarcoma and Synovial sarcoma.
Parosteal osteosarcoma (PO) is a well-differentiated low-grade malignant bony tumour (Kumar, Barwar, and Khan 2014). It is the most common subtype of surface osteosarcoma. PO is characterized as a slow growing bony mass with a “cauliflower-like” appearance. On radiography, PO usually presents as a radiodense lesion arising from the metaphyseal region of long bones (Prabowo et al. 2020), and may present with a “string sign”: a thin, radiolucent line between the tumour and the bone cortex (Idrees, Zarrar, and Mujtaba 2023). In most findings, the lesion is poorly demarcated with calcifications. Studies have observed that in osteosarcoma, calcification begins at the center and spreads towards the periphery, unlike in MO where it begins at the periphery and progresses inwards (Man et al. 2011). In our case, peripheral calcification can be seen on radiography and MRI. Moreover, amplification of the MDM2 gene, an oncogene commonly associated with low-grade osteosarcoma and recognized as an effective diagnostic marker for this osteosarcoma (Dujardin et al. 2011) (sensitivity = 81.3%, specificity = 100%) (L. Li et al. 2024), was also absent in our patient. Both findings made a diagnosis of osteosarcoma very unlikely.
Synovial sarcoma is a malignant slow-growing soft tissue tumour that can arise from anywhere in the body (Saad et al. 2021). Most SS patients present with a prolonged history of a painless slow-growing mass, with an average duration of 2-4 years before seeking medical assistance. Conversely, Patients with MO usually seek clinical evaluation a few months after the occurrence, presenting with pain due to the inflammatory process during the early stage as mentioned above. On radiography, approximately 30% of SS cases present with “fine, stippled” calcification (Wilkerson et al. 2012). Calcifications in SS are predominantly situated at the periphery, resembling the pattern observed in MO. Bony erosion in synovial sarcoma can also occur when the tumour is located in close proximity to bone structures, though it is observed in only about 22% of cases (Luczyńska et al. 2014). On the other hand, MO never presents with bony erosion (Luczyńska et al. 2014). In our patient, the lesion was closely situated near the left clavicle yet no bony erosion was observed. Given that our patient had no bony erosion and presented with a fast-growing painful mass, a diagnosis of SS was unlikely. Granted, more gene expression studies (Transducin-Like Enhancer 1 (TLE1)) can be done to definitively exclude SS (El Beaino et al. 2020).
In our case, histopathological and molecular findings, correlated with imagery findings and clinical history, were used to arrive at a diagnosis of MO. There were immature woven bone formations, coupled with no signs of significant cytological atypia. Ideally, the presence of progressive maturation of the lesion from the centre outwards, known as “zoning” phenomenon, should be used as a diagnostic feature (Nishio et al. 2010). However, due to the nature of the biopsy (incisional), we were unable to observe this. Despite this, the detection of a COL1A1::USP6 gene fusion in molecular testing provides substantial evidence supporting a diagnosis of myositis ossificans (MO), given its established association with MO in several case series. In one study, COL1A1-USP6 rearrangement was identified in 5 out of 7 cases of MO (Švajdler et al. 2019), while another series reported USP6 rearrangements in 8 of 9 MO cases (Bekers et al. 2018), reinforcing this genetic alteration as a significant diagnostic marker for MO.
In terms of treatment, there are two main approaches: surgical excision and conservative radiographic monitoring. Studies have recommended surgical excision of MO in symptomatic mature lesions after at least six months (Cherry et al. 2023). Surgical excision of MO during the early phase can lead to reoccurrence (Cherry et al. 2023), and is therefore not indicated in our case. Given MO’s self-limiting nature, a majority of MO are treated conservatively (Saad et al. 2021). Apart from continual radiographic monitoring to ensure the spontaneous resolution of the mass, NSAIDs can stop further progression of MO and decrease inflammation (Park et al. 2024). As for our patient, indomethacin was prescribed along with continual radiographic monitoring and he recovered without complications.
A review of literature was also done to document existing cases of atraumatic MO in the paediatric population (<18 years old). With reference to a systematic scoping review of MO in the pediatric population done in 2022 (Cherry et al. 2023), as well as a search on PubMed database, a total of 22 cases of atraumatic MO in the paediatric population were found from 2004 to 2024.
From the literature review, it is noted that biopsy was done in 13 out of 22 cases (Percutaneous: 7, 31.8%; Incisional: 4, 18.2%; Excision: 2, 9.1%; None: 6, 27.3%; Not Documented: 3, 13.6%) as imagery studies were unable to sufficiently exclude malignancy. As for treatment, 8 out of 22 cases (36.4%) adopted a conservative approach while 13 out of 22 cases (59.1%) opted for excision instead.
In conclusion, tumours presenting as atraumatic MO should raise a high clinical suspicion for malignancy and warrant thorough investigation. In this case, plain radiography and clinical examination were insufficient to exclude soft tissue malignancy due to clinical similarities between MO and sarcomas, and the absence of a history of trauma. Biopsy and molecular studies are recommended for accurate diagnosis and appropriate management when imaging investigation remains inconclusive. Early and comprehensive evaluation is essential to ensure timely treatment and improve patient outcomes.
ACKNOWLEDGMENT
Author gracefully acknowledges contributions from colleagues and willful participation from patient.
FINANCIAL SUPPORT AND SPONSORSHIP
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
INFORMED CONSENT
Consent was obtained from the patient for the publication of the case report and any accompanying images.